

We are ready to help you overcome the challenges of producing and developing innovative biotechnological products and advancing scientific research

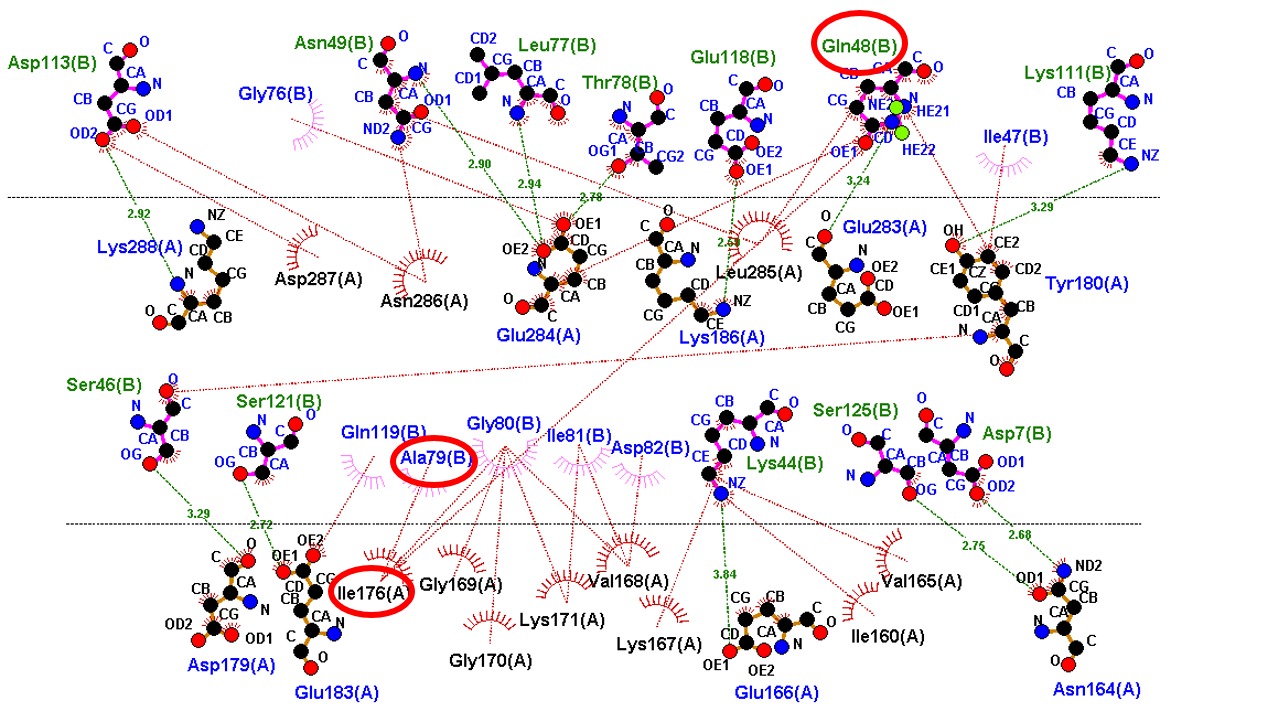
Protein-Protein Docking (PPD) is an essential computational tool that extends our expertise beyond single-enzyme modification. We utilize PPD to model the complex interactions between our engineered enzymes and other molecular components. This allows us to rationally design multi-enzyme complexes with optimal orientation for enhanced production efficiency, predict and prevent undesirable protein aggregation, and ensure our biocatalysts are fully integrated into demanding industrial systems. PPD is key to engineering stable, functional, and highly efficient biochemical systems.
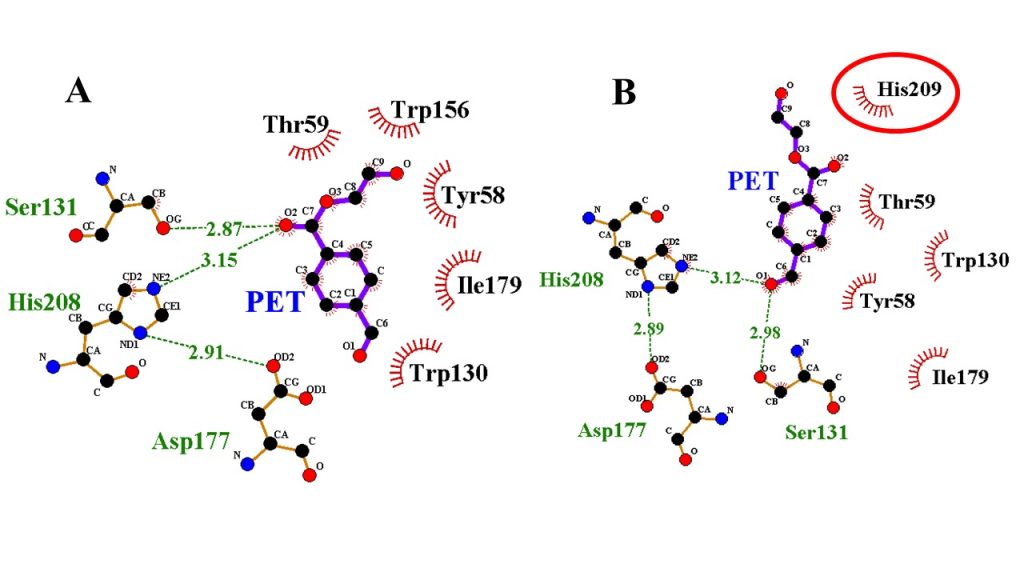
As part of our commitment to rational enzyme engineering, we utilize Site-Specific Molecular Docking as a critical computational tool. This advanced approach focuses the simulation exclusively on a pre-defined active site or binding pocket within the enzyme’s structure. By restricting the search area to key catalytic regions, we dramatically increase the speed and accuracy of predicting how a specific substrate or inhibitor will interact. This function is essential for our work in designing mutant enzymes with enhanced specificity, determining the optimal fit for novel bio-based products, and ensuring that the structural modifications we engineer will achieve the desired catalytic outcome.
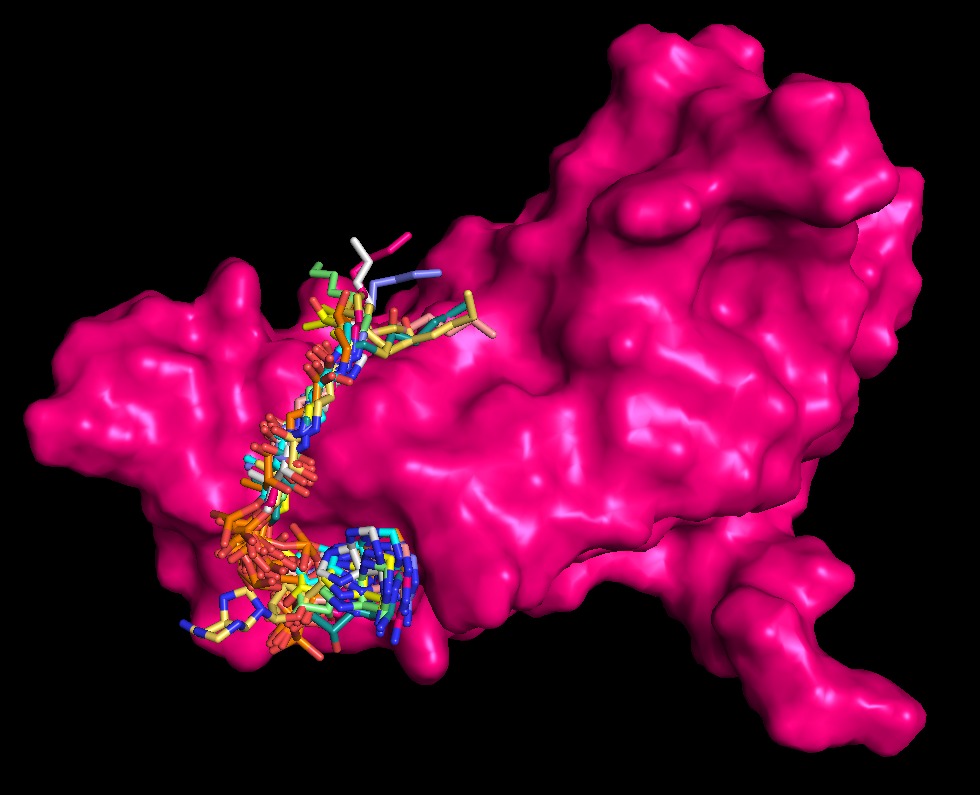
We elevate our initial screening capabilities by employing Blind Molecular Docking powered by advanced AI tools like DiffDock. Unlike traditional docking, DiffDock uses generative diffusion models to rapidly sample and accurately predict binding poses and affinities across the entire enzyme surface, even for previously unknown ligands or binding sites. For Smart Protein Builders, this method is instrumental in the early stages of our rational enzyme engineering pipeline, enabling us to quickly identify novel allosteric sites (hidden regulatory pockets) or unexpected off-target interactions. This proactive, AI-driven insight ensures our optimized enzymes possess maximal specificity and stability by accounting for the full landscape of potential molecular interactions.
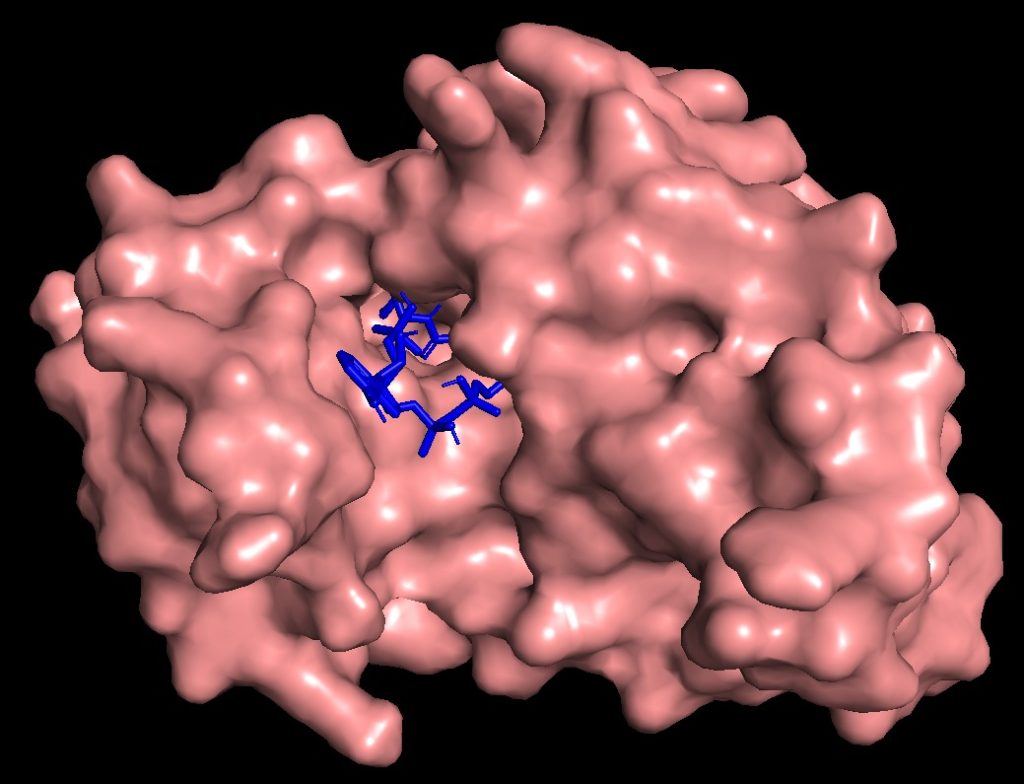
Molecular Docking is a core computational technique we employ to predict the optimal binding pose and affinity between an enzyme (receptor) and a substrate or inhibitor (ligand). This method simulates the molecular interactions, allowing us to accurately score and rank potential binding partners before costly lab synthesis. By providing critical insights into the strength and nature of the protein-ligand complex, docking is essential for hit identification in drug design, virtual screening for novel inhibitors, and the rational design of mutant enzymes with enhanced specificity or catalytic function. We utilize advanced docking algorithms to ensure high precision in predicting molecular recognition.
+98 912 836 0916
+98 930 144 1004
nimanezhad86@gmail.com


Custom-optimized rational enzyme engineering for your process requirements
©۲۰۲5 .neoenzyme all rights reserved